Introduction
Ewing Sarcoma (ES) is a malignant primitive neuroectodermal tumor within the small round cell tumor category. It is cytogenetically characterized by the t(11;22)(q24;q12) translocation, resulting in an EWSR1-FLI1 fusion. ES is the second most frequent primary malignant bone tumor in children and adolescents, and much rarer in adults. Its low incidence in the adult population has led to a paucity of specific studies in this group, limiting knowledge regarding its clinical behavior and prognosis.
Clinically, ES typically presents with localized pain, a palpable mass, or symptoms related to compression of adjacent structures. Due to its aggressive nature and propensity for hematogenous spread, a multimodal treatment approach is essential. This involves intensive systemic chemotherapy in combination with local treatments such as surgery, radiotherapy, or both. The introduction of chemotherapy regimens like VAC/IE has significantly improved outcomes, especially in pediatric patients with localized disease.
Despite pediatric patients achieving 5-year survival rates of up to 70% in localized disease, adults often experience worse outcomes. This may be due to lower treatment tolerance and a higher incidence of adverse prognostic features such as larger tumor size, axial location, or non-resectability, along with patient-related factors including comorbidities.
Materials and methods
This study aimed to analyze oncologic outcomes and identify prognostic factors associated with survival in adult ES patients treated at a national reference center, and to compare these findings with pediatric cohorts.
A retrospective observational study was conducted at the CSUR at HGUGM. Thirty-three patients over 20 years of age, diagnosed with ES between 2015 and 2022, were included. Clinical data were collected from medical records up to March 2024. Diagnosis was confirmed through histopathology and, when required, molecular techniques.
Twelve variables were recorded, including: age, sex, tumor location (peripheral vs. central), primary site (osseous vs. extraosseous), tumor size (<8 cm vs. ≥8 cm), metastatic status at diagnosis (localized vs. metastatic), type of local treatment (surgery, RT, or both), chemotherapy use, and surgical margin status (R0, R1, R2). OS and DFS were also evaluated (Table 1).
Univariate analyses used chi-square or Fisher’s exact tests for categorical variables and the Wilcoxon test for continuous variables. Multivariate analysis was performed using Cox regression to identify independent prognostic factors. Odds Ratios (ORs) and 95% Confidence Intervals (CIs) were calculated.
OS and DFS were estimated using Kaplan-Meier curves, and log-rank tests were used for comparisons. Statistical analysis was conducted using IBM SPSS Statistics v30. A p-value <0.05 was considered statistically significant.
The study was approved by the institutional ethics committee. Given the retrospective nature of the study, individual informed consent was waived.
Results
Descriptive analysis
The cohort included 33 adult patients with ES, with a mean age of 37 years (range: 20-84, SD: 17.02); 67.9% were male. Anatomically, 50% of tumors were located in the limbs, 28.6% in the pelvic girdle, 17.9% in the spine, and 3.6% in the shoulder girdle. Most cases (78.8%) were osseous in origin, while 21.2% were extraosseous.
At diagnosis, 63.3% of patients had localized disease, and 36.4% presented with metastases. Tumor size was <8 cm in 76.9% and ≥8 cm in 23.1% of patients.
Regarding treatment, surgery was performed in 33.3%, RT in 18.2%, and combined surgery + RT in 30.3%. Local treatment was not administered in 18.2% of patients due to advanced disease and irresectability. Chemotherapy was given to 93.9% of patients. Among those undergoing surgery, 81.3% achieved R0 margins, 12.5% had R1 margins, and 3.6% had R2 margins.
Univariate and multivariate analysis
In univariate analysis, metastatic disease at diagnosis (p=0.04; OR: 8.3; 95% CI: 1.3-51.6) and age ≥35 years (p=0.019; OR: 3.8; 95% CI: 1.2-19.8) were significantly associated with worse OS. Other variables such as sex, location, tissue type, and local treatment did not reach statistical significance (Table 2). In multivariate analysis, only the presence of metastases remained significant (p=0.025; OR: 4.7; 95% CI: 1.56-14.24). Age ≥35 showed a trend but was not significant (p=0.072) (Table 3).
Survival analysis
With a median follow-up of 34 months (range: 2.5-152), the 5-year OS was 44%, and 5-year DFS was 41%. Patients without metastases had a mean survival of 85 months, compared to 28 months in those with metastases. The 5-year OS was 73% in localized disease versus 20% in metastatic disease. During follow-up, 39.4% experienced recurrence, and 48.5% died.
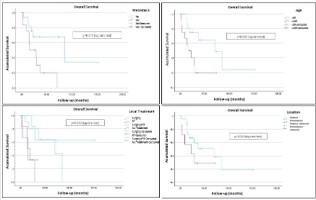
Kaplan-Meier analysis showed significantly reduced OS in patients with metastases at diagnosis (p=0.015) and those aged ≥35 years (p=0.04), though the latter lost significance in multivariate analysis (Figure 1). DFS was significantly lower in patients with soft tissue tumors compared to osseous origin (p=0.005). Combined local treatment showed a trend toward improved OS, while lack of local treatment correlated with the poorest outcomes (Figure 2).
Table 1: Studied variables.
| Variables |
Number (Median) |
% [Range] |
| Sex |
| Male |
22 |
667 |
| Female |
11 |
33.3 |
| Age |
-37 |
[20,84] |
| <35 |
18 |
54.5 |
| ≥35 |
15 |
45.5 |
| Tumor location |
| Peripheral |
14 |
42.4 |
| Central |
19 |
57.6 |
| Primary Site |
| Osseous |
26 |
78.8 |
| Extraosseous |
7 |
21.2 |
| Tumor size |
| <8 |
20 |
76.9 |
| >8 |
6 |
23.1 |
| Mestastatic |
| No |
21 |
63.6 |
| Yes |
12 |
36.4 |
| Local Treatment |
| Surgery |
11 |
33.3 |
| RT |
6 |
18.2 |
| Surgery + RT |
10 |
30.3 |
| No local treatment |
6 |
18.2 |
| Surgical margins |
| R0 |
18 |
87.3 |
| No R0 |
3 |
12.7 |
| Chemotherapy |
| Yes |
31 |
93.9 |
| No |
2 |
6.1 |
| Recurrence |
13 |
39.4 |
| Complete remission |
13 |
39.4 |
| Deaths |
16 |
48.5 |
Table 2: Univariate analysis.
| |
Overall |
Survivle |
Disease-Free |
Survivle |
| |
p |
OR (95% IC) |
p |
OR (95% IC) |
| Stage |
| No Metastatic |
0.015 |
8.3(1.3-51.6) |
NS |
2(0.469-8.53) |
| Metastatic |
| Age |
| <35 |
0.04 |
3.8(1.2-19.8) |
NS |
1.75(0.42-7.1) |
| >35 |
| Primary Site |
| Osseous |
NS |
2.3(0.4-14.5) |
NS |
1.2(0.22-6.52) |
| Extraosseous |
| Tumor Size |
| <8 cm |
NS |
2.3(0.3-16.5) |
NS |
1.5(0.24 - 9.4) |
| 8 cm |
Table 3: Multivariate analysis.
| |
Overall |
Survivle |
| |
p |
OR (95% IC) |
| Stage |
| No Metastatic |
0.025 |
4.7(1.56-14.24) |
| Metastatic |
| Age |
| <35 |
NS |
3.2(0.9-11.4) |
| >35 |
| Primary Site |
| Osseous |
NS |
1.4(0.4-5.2) |
| Extraosseous |
Discussion
Our results demonstrate that Ewing sarcoma in adults follows a clinical course similar to that reported in the literature, with lower OS and DFS rates compared to pediatric populations. In our series, the 5-year OS was 43.9%, compared to over 65% in pediatric studies. This discrepancy becomes more pronounced when comparing patients without metastases (73%) to those with metastases (20%), reinforcing metastatic disease at diagnosis as a critical adverse prognostic factor.
Studies such as Karski et al. confirm that although adults with localized disease may achieve survival rates comparable to pediatric patients, outcomes are significantly worse in metastatic settings. Esiashvili et al. and Dirksen et al. have also documented improved pediatric survival over time, in contrast with stagnation in adults. Other studies by Leavey, Gupta, and Pretz highlight consistent differences between children and adults, even in similar localized disease contexts.
In our cohort, only metastatic disease at diagnosis remained significant in multivariate analysis. This aligns with the findings of Sánchez Pérez et al., further solidifying it as the primary poor prognostic factor. Krakorová et al. suggested complete remission as a stronger predictor than metastatic status. Similarly, Cotterill, Bacci, and Ahmed have emphasized chemotherapy response as a key determinant of DFS.
While other unfavorable factors such as tumor size and axial location have been identified (e.g., Duchman, Karski, Jawad), these did not reach significance in our analysis. Additional factors, including race, socioeconomic status, and comorbidities, have been linked to diagnostic delay and poorer outcomes. Adults’ lower tolerance to intensive chemotherapy regimens and increased incidence of unresectable or bulky tumors likely contribute to their worse prognosis.
Local treatment choice also influenced prognosis. Patients receiving combined surgery and RT had the best outcomes, suggesting this strategy should be prioritized when feasible. Furthermore, the sharp drop in DFS in extraosseous tumors points to greater biological aggressiveness and possibly differential therapeutic responses.
Verma et al. demonstrated that timely, combined surgery and RT, along with chemotherapy, improved survival. Histological response to neoadjuvant chemotherapy has also been strongly associated with DFS, as shown by Bacci, Lee, and the Memorial Sloan Kettering Cancer Center. Gupta, the Polish Sarcoma Group, and the National Cancer Database suggest that if tolerated, adults may benefit from pediatric-inspired protocols.
Limitations
Our study has limitations. Its retrospective design may introduce bias in data collection and interpretation. The sample size is limited, reducing statistical power, especially in multivariate analysis. Molecular biomarkers and treatment toxicity were not evaluated, though these may influence outcomes and treatment tolerability in adults.
Nevertheless, this study contributes valuable data on an underrepresented population in the literature, offering updated insights into adult ES in Spain. It confirms the prognostic importance of metastatic disease and highlights the relevance of chemotherapy response, comorbidity management, and strategic treatment selection in improving outcomes.
A multidisciplinary approach, integrating optimal systemic and precise local therapies, may significantly enhance prognosis in adult patients and approach the outcomes seen in pediatric cohorts when diagnosed early.
Conclusions
Ewing sarcoma in adults is associated with poorer prognosis compared to pediatric cases, particularly when metastatic at diagnosis. Our findings reinforce the need for early diagnostic strategies and adult-specific therapeutic protocols to intervene before disease dissemination.
Future prospective studies and personalized treatment strategies are essential to address the complexities of managing adult ES. Larger, multicenter studies are required to establish definitive therapeutic recommendations and improve outcomes in this patient population.
Declarations
Conflicts of interest: The authors declare no conflicts of interest.
Funding: This study was funded by Project TED2021-132200BI00 (Strategic Projects for Digital Transition 2021, State Research Agency, Ministry of Science and Innovation).
This study was also funded by Instituto de Salud Carlos III (ISCIII) through the Biomodels and Biobanks Platform and co-funded by the European Union (PT23/00116 to RPM).
Acknowledgments: We thank the CSUR sarcoma team at HGUGM for their support in data collection, as well as the ethics committee for study approval.
Abbreviations: CSUR: National Reference Sarcoma Center; HGUGM: Gregorio Marañón General University Hospital; 95% CI: 95% Confidence Interval; OR: Odds Ratio; RT: Radiotherapy; ES: Ewing Sarcoma; OS: Overall Survival; DFS: Disease-Free Survival; VAC/IE: Chemotherapy regimen (Vincristine, Adriamycin, Cyclophosphamide/Ifosfamide, Etoposide).
References
- Ewing J. Diffuse Endothelioma of Bone. Proc N Y Pathol Soc. 1921; 21: 17–24.
- Jaffe N, Traggis D, Cassady JR, et al. Improved outlook for Ewing’s sarcoma with combination chemotherapy. Cancer. 1976; 38: 1925–1934.
- Gaspar N, Hawkins DS, Dirksen U, et al. Ewing Sarcoma: Current Management and Future Approaches Through Collaboration. J Clin Oncol. 2015; 33: 3036–3046.
- American Cancer Society. Ewing Tumor Survival Rates. 2024. Available at: https://www.cancer.org/cancer/types/ewing-tumor/ detection-diagnosis-staging/survival-rates.html
- Verywell Health. What Is Ewing Sarcoma? 2023. Available at: https://www.verywellhealth.com/ewing-sarcoma-7481395
- PubMed. Treatment differences and long-term outcomes in adults and children with Ewing sarcoma. 2024. Available at: https:// pubmed.ncbi.nlm.nih.gov/39179493/
- Leavey PJ, Collier AB. Ewing sarcoma: prognostic criteria, outcomes and future treatment. Expert Rev Anticancer Ther. 2008; 8: 617–624.
- Gupta AA, Pappo A, Saunders N, et al. Clinical outcomes of children and adults with localized Ewing sarcoma. Cancer. 2010; 116: 3189–3194.
- Duchman KR, Gao Y, Miller BJ. Prognostic factors for survival in patients with Ewing’s sarcoma using the SEER program database. Cancer Epidemiol. 2015; 39: 189–195.
- Krakorová K, Jančářová A, Krejčíříková H, et al. Advantages in prognosis of adult patients with Ewing sarcoma. Clin Transl Oncol. 2021; 23: 34–41.
- Karski EE, McIlvaine E, Segal MR, et al. Identification of discrete prognostic groups in Ewing sarcoma. Pediatr Blood Cancer. 2016; 63: 47–53.
- Cotterill SJ, Ahrens S, Paulussen M, et al. Prognostic factors in Ewing’s tumor of bone. J Clin Oncol. 2000; 18: 3108–3114.
- Jawad MU, Cheung MC, Min ES, et al. Ewing sarcoma demonstrates racial disparities in incidence-related and sex-related differences in outcome. Cancer. 2009; 115: 3526–3536.
- Lee JA, Kim DH, Lim JS, et al. Soft tissue Ewing sarcoma: characteristics and outcomes of extraosseous Ewing sarcoma in pediatric and adult populations. Int J Clin Oncol. 2016; 21: 963–970.
- Esiashvili N, Goodman M, Marcus RB. Changes in incidence and survival of Ewing sarcoma patients over the past 3 decades. J Pediatr Hematol Oncol. 2008; 30: 425–430.
- Bacci G, Longhi A, Ferrari S, et al. Histologic response of nonmetastatic Ewing’s sarcoma family tumors to neoadjuvant chemotherapy correlates with clinical outcome. J Chemother. 2004; 16: 293–300.
- Sánchez Pérez C, Mediavilla Santos L, Carbó Laso E, et al. Are we able to predict the outcome of Ewing sarcoma? Arch Clin Exp Surg. 2017.
- Verma V, Lin SH, Simone CB. The influence of treatment timing and modalities on outcomes in Ewing sarcoma: A population-based study. J Natl Compr Canc Netw. 2018; 16: 706–715.
- Pretz JL, Taggart DR, Dombi E, et al. Ewing Sarcoma in Adults: Survival and Treatment Outcomes in the Era of Modern Chemotherapy. Oncologist. 2017; 22: 1436–1443.
- Ahmed SK, Robinson SI, Rose PS, et al. Adult Ewing Sarcoma: Survival and Local Control Outcomes in the Modern Era. Sarcoma. 2013; 2013: 268016.
- Striefler JK, Donati D, Bhangu JS, et al. The impact of comorbidities on overall survival in adult patients with Ewing family tumors: a multicenter study. Cancer Med. 2022; 11: 4171–4180.
- Dirksen U, Brennan B, Le Deley MC, et al. High-Dose Chemotherapy Compared With Standard Chemotherapy and Lung Radiation in Patients With Primary Disseminated Ewing Sarcoma: Results of Euro-E.W.I.N.G. 99 and Ewing 2008. J Clin Oncol. 2018; 36: 3182–3190.
- Schuck A, Ahrens S, Paulussen M, et al. Radiotherapy in Ewing’s sarcoma: an analysis of 1988 patients treated on the CESS 81, CESS 86, and EICESS 92 trials. Int J Radiat Oncol Biol Phys. 2003; 55: 168–177.
- Raciborska A, Bilska K, Drabko K, et al. Clinical characteristics and outcome of adult patients with Ewing sarcoma: report of the Polish Sarcoma Group. J Clin Med. 2021; 10: 3627.
- Lee RJ, Gittleman H, Barnholtz-Sloan JS, et al. Incidence and survival of malignant bone tumors in the United States: National Cancer Database analysis. Eur J Med Res. 2023; 28: 13.